A key part of this project is to parse the secondary structure line of Stockholm files so that it can be interpreted for coloring schemes. I have been adding mini-goals as appropriate. I will probably also need to add code to check that the sequence length and secondary structure length are the same, as well as the same number of open and closed parentheses.
WUSS notation is used in RNA stockholm files to indicate secondary structure. WUSS notation can support more characters than I thought, but
Rfam uses the simplified version that the covariance modeling program
Infernal uses. The description of Rfam on the
Janelia Farm page is
Rfam is a collection of multiple sequence alignments and covariance models covering many common non-coding RNA families. The main use of Rfam is as a source of RNA multiple alignments with consensus secondary structure annotation in a consistent format. In conjunction with the Infernal software package, Rfam covariance models (CMs) can be used to search genomes or other DNA sequence databases for homologs to known structural RNA families.
WUSS notation uses <>, (), [], and {} to indicate base pairs and ':', ',', '_', '.', and '~' as single stranded columns. Each type of symbols has subtle meaning, but for Infernal the structure annotation line only needs to indicate which columns are base paired to each other. Thus, full WUSS notation is not necessary and a simple minimal annotation uses <> to indicate base pairs and '.' for single stranded positions of the alignment.
In more detail taken from the Infernal user guide:
Base pairs: the different symbols indicate different depth
*<> for simple terminal stems
*() for "internal" helices enclosing a multifunction of all terminal stems
*[] for internal helices enclosing a multifunction that includes at least one annotated () stem already
*{} for all internal helices enclosing deeper multifurcations
Hairpin loops
*indicated by underscores '_'
*Simple stem loops example: <<<____>>>
Bulge, interior loops
*indicated by dashes '-'
Multifurcation loops
*indicated by commas ','
*example: <<<___>>>,,<<<__>>>
External residues completely outside structure
*indicated by colons ':'
Insertions
* . to a known structure
* ~ used to indicate that a local structural alignment left regions of target and query unaligned.
Pseudoknots
* pairs of upper case/lower case letters
* example: <<<<_AAAA____>>>>aaaa
Things that I am thinking about:
-I need to interpret WUSS notation in a general way. It shouldn't be too difficult, but it is necessary since the same structure can be written in multiple ways. An example from the Infernal user guide is : <<<<....>>>> and ((((____)))) and <(<(._._)>)> all indicate a four base stem with a four base loop
-How should I store the secondary structure line so that it will be easily interpreted to implement coloring schemes?
Potentially I can store pairs of positions like how the disulfide bond positions are stored as annotations (Jim pointed this one out). I also need to keep in mind that bulges might exist, so I can't just interpret a run of the same type of bracket as part of the same stem. VARNA interprets bulges just fine, so I don't have to worry about that. An example of a complicated structure with a bulge:
<<<<……<<<< <<<<…..>>>>..>>>>……<<<<…>>>>….>>>>
-How can I make sure that there are 4 stems instead of 3? I can't simply scan through from left to right or eat away at both ends at the same time. It looks like the
RALEE mode for Emacs handles bulges just fine based on this example in the readme
0123456789012345678901234
.<<<<<...>>.<<...>>..>>>.
Column 1 pairs with 23
2 with 22
3 with 21
4 with 10
5 with 9
12 with 18
13 with 17
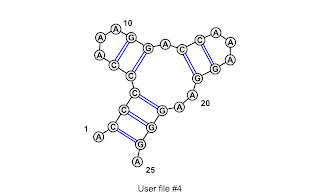
The image is from VARNA. Note the numbering in the image starts at 1 instead of 0.
RALEE is written in Emacs Lisp, so I need to look up some basics in Lisp before I can feel confident that I'm interpreting the code correctly! I think that this code will cut down on my thinking time, however.
To do
-check that secondary structure line and sequence are the same length. Does Jalview already do this?
-change all bracket types to () for VARNA (I just noticed that VARNA only likes (), not <> for base pairing! )
-convert all WUSS symbols to something simple, like how Jalview already does for protein secondary structure (simple helices and sheets)
-Need to figure out how to detect pseudoknots
-Add support for error checking when a user adds a base pair annotation. Make sure same number of column groups are selected
-How will colors cycle for different numbers of stems?

















